Ziel der Übung ist die Quantifizierung von Proteinen mittels photometrischer Bestimmungsmethoden, wobei die verwendeten Methoden bezüglich Empfindlichkeit, Nachweis-, Erfassungs- und Bestimmungsgrenze verglichen werden sollen. Für eine ausgegebene Proteinlösung unbekannter Konzentration soll die geeignete Kalibrierfunktion für die Berechnung des Proteingehaltes gewählt werden. (Prüfung: Aufbau eines Proteins!)
Spektrometrische Verfahren beruhen darauf, dass Lichtquanten bestimmter Energie stoffspezfisch absorbiert werden. Allgemein gilt, dass elektromagnetische Strahlung von Molekülen absorbiert wird. Die Gesetzmässigkeit der Absorption ist durch das Lambert-Beersche Gesetz beschrieben:

Bezieht man sich auf verdünnte Lösungen (c < 10-2 mol L-1) bei denen ausschliesslich der gelöste Stoff der Konzentration c absorbiert, folgt daraus:
𝐸 =log5 6=log+, += 𝜀 𝑐 𝑑 ... Lambert-Beer’sches Gesetz
D.h. die Absoprtion ist direkt proportional (ausgedrückt durch ε) zur Wegstrecke d (cm) in einer Lösung sowie zur Konzentration c (mol L-1) der Lösung.
| T | ... | Transmission (Durchlässigkeit), e |
| O.D. | ... | Opazität (optische Dichte = O.D., engl.: Optical density) |
| E | ... | Extinktion |
| I | ... | Intensität eines monochromatischen Lichtstrahls |
| c | ... | Konzentration (mol L-1) |
| d | ... | Wegstrecke (cm) |
| α | ... | Absorptionskoeffizient |
| ε | ... | molarer dekadischer Extinktionskoeffizient (mol-1 cm-1 L) |
Spektroskopische Messungen können in unterschiedlichen Wellenlängenbereichen durchgeführt werden, photometrische Messungen meist im UV und VIS Bereich:
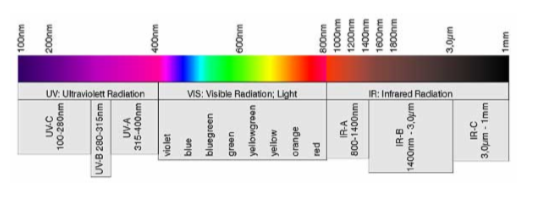
Wenn die Eigenabsorption der zu untersuchenden Verbindung nicht ausreicht, um niedrige Konzentrationen verlässlich zu messen, müssen chemische Reaktionen durchgeführt werden. Diese Reaktionen führen zu Verschiebungen der Absorptionsmaxima zu höheren oder tieferen Wellenlängen.
Proteine zeigen an der Polypeptidkette und durch ihre prosthetischen Gruppen Absorption. Die Peptidbindung selbst führt zu einem Absorptionsmaximum bei 190 nm. Das zweite Absorptionsmaximum der Peptidbindung, bei ca. 220 nm, ist oftmals durch Absorption der Aminosäureseitenketten überlagert. Seitenketten der aromatischen Aminosäuren (Phenylalanin λmax 260 nm, Tyrosin λmax 275 nm, Tryptophan λmax 280 nm) geben eine ausgeprägte Absorption bei 260 – 280 nm. Der Hauptbeitrag stammt von der Aminosäure Tryptophan. Durchschnittliches Vorkommen der wichtigsten UV absorbierenden Aminosäuren in Proteinen: Phenylalanin 3.9 %, Tyrosin 3.2 %, Tryptophan 1.3 %.
Diese Absorption kann zur einfachen, wenn auch nicht sehr genauen, Konzentrationsbestimmung herangezogen werden.
Messung bei 280 nm: Die photometrische Bestimmung ist durch UV absorbierende Substanzen (Pigmente, phenolische Verbindungen, organische Kofaktoren) beeinträchtigt. So tragen auch Nukleinsäuren zu einer Absorption bei 280 nm bei. Je nach verwendetem Gerät können Proteinkonzentrationen von 20-3000 µgmL-1 eingesetzt werden.
Messung bei 205 nm: Proteinkonzentrationen von 1-100 µgmL-1 können gemessen werden, wobei hier alle Substanzen mit C=C oder C=O Doppelbindungen störend wirken, sowie geringe Verunreinigungen in den verwendeten Pufferkomponenten, die wiederum nicht hoch konzentriert sein dürfen.
Der Name dieser Methode beruht auf einer Farbreaktion von gelöstem Biuret (Carbamoylharnstoff NH2-CO-NH-CO-NH2) und Kupfersulfat in alkalischem, wässrigem Milieu (Biuret – Reaktion). Es entsteht ein rotvioletter Farbkomplex zwischen den Cu2+ Ionen und je 2 Biuretmolekülen. Diese Farbreaktion ist typisch für Verbindungen mit mindestens 2 CO-NH-Gruppen (Peptidbindungen) und kann daher auch für den kolorimetrischen Nachweis von Peptiden und Proteinen verwendet werden.
Der farbige Protein-Cu2+-Komplex, der bei der Biuret-Reaktion entsteht:
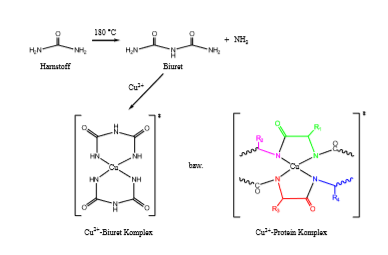
Sind Tyrosinreste vorhanden, tragen diese ebenfalls merklich - durch die Komplexierung von Kupferionen - zur Farbstoffbildung bei. Die Messung des Farbkomplexes kann bei ca. 550 nm erfolgen.
Störend wirken vor allem Ammonium, schwach reduzierende und stark oxidierende Substanzen (Sulfhydrylverbindungen, Natriumphosphat, Glucose, Ammoniumsulfat). Schwache Konzentrationen an Sodiumdodecylsulfat (SDS) oder anderen Detergentien sind hingegen tolerabel.
Der Biuret-Assay ist im Vergleich zu anderen Assays der unempfindlichste. Nachweisegrenzen (geräteabhängig): ca 1-10 µgmL-1.
Unter alkalischen Bedingungen bindet Cu2+ an den Stickstoff der Amidbindungen der Proteine (Biuret-Reaktion). Dieser Komplex wird im Lowry-Assay, einer Weiterentwicklung des BiuretAssays, durch Na-K-Tartrat stabilisiert.
Dieser Komplexreaktion folgt eine Reduktion des verwendeten Folin-Ciocalteau-Phenol Reagenzes im alkalischen Milieu. Kupfer unterstützt hier den effektiven Elektronentransfer auf die Chromophore dadurch, dass es das Peptidgerüst stabilisiert.
Folin-Ciocalteau-Phenol Reagens: Na2WO4 Natriumwolframat, Na2MoO4 Natriummolybdat, 85% Phosphorsäure, conc. HCl, Li2SO4 Lithiumsulfat und "einige Tropfen" Br2 (verkocht) reagieren zu hexavalenter Phosphorwolfram- und Phosphormolybdänsäure: 3H2O × P2O5 × 13WO3 × 5MoO3 × 10H2O , 3H2O × P2O5 × 14WO3 × 4MoO3 × 10H2O.
Das Reagenz enthält somit 6-wertiges Wolfram und Molybdän welche durch polare Aminosäuren, Cystein, Histidin, Tyrosin oder Tryptophan soweit reduziert werden, dass farbige Mischoxide von 4- bis 6-wertigen Metallen entstehen.
Ohne Kupferzugabe wäre die Farbintensität primär durch Tyrosin und Tryptophan bestimmt. Li2SO4 beugt der Bildung von unlöslichen Na-Salzen (Trübung) vor. Die resultierende tiefblaue Färbung wird bei einer Wellenlänge von 750, 650 oder 550 nm gemessen.
Anmerkung: Das Reagens selbst enthält kein Phenol! Der Name kommt daher, dass dieses Reagens mit Phenolen und nicht-phenolischen, reduzierenden Verbindungen Chromogene bildet, die spektroskopisch detektierbar sind.
Chromogen – Chromogene sind chemische Verbindungen, die selbst nicht farbig sind, aber chromophore Gruppen bilden Chromophor – Bezeichnung für Atomgruppierungen, die einer Verbindung durch selektive Lichtabsorption "Farbigkeit" verleihen; im allgemeinen handelt es sich bei den chromophoren Gruppen um π-Elektronensysteme
Die Lowry Methode ist eine der höchst sensitivsten und die am häufigsten verwendete Methode für die quantitative Proteinbestimmung. Diese photometrische Methode erlaubt es, geräteabhängig Proteinkonzentrationen von 1-100 µgmL-1 zu detektieren Allerdings ist diese Methode einigen Limitierungen unterworfen: Vor allem EDTA, GuanidinHCl, Triton X-100, SDS, Brij 35, > 0.1M Tris, 1M Natriumacetat, 1 M Natriumphosphat, Ammonsulfatkonzentrationen >5%, Glycin und auch reduzierende Agentien stören die Bildung des Farbkomplexes.
In Gegenwart von Proteinen verschiebt sich das Absorptionsmaximum von Coomassie Brillantblue G250 (blauer Säurefarbstoff) im sauren Milieu von 465 nm nach 595 nm. Die Zunahme der Absorption bei 595 nm, die mit einem Farbumschlag von rot nach blau einhergeht, entsteht durch die Stabilisierung des Farbstoffes in seiner unprotonierten, anionischen Sulfonatform durch Komplexbildung zwischen Farbstoff und Protein, die mit der zu bestimmenden Proteinkonzentration korreliert.
Coomassie Brillian Blue G-250:
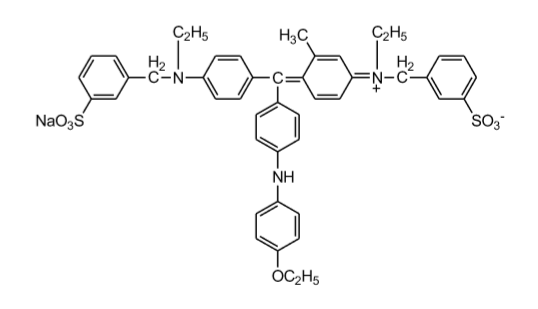
Der Farbstoff bindet unspezifisch (nicht-kovalent) an kationische und nichtpolare, hydrophobe Seitenketten der Proteine, wobei er in seiner Sulfonatform stabilisiert wird. Am wichtigsten ist dabei die Wechselwirkung mit Arginin, aber auch Lysin, Histidin, Tryptophan, Tyrosin und Phenylalanin tragen zur Komplexstabilisierung bei.
Diese Methode ist um den Faktor 2-5 sensitiver als die Lowry-Methode, die Nachweisgrenze liegt zwischen 0.05-0.5 µgmL-1 (geräteabhängig). Großer Vorteil: nur wenige Substanzen stören die Komplexbindung. z.B. Natriumdesoxycholat, hohe Konzentrationen an Triton X-100 (> 0,5 %) und größere SDS Konzentrationen (> 0,1 %). Vor Allem aber ist die Methode tolerant gegenüber Reduktionsmitteln.
Ein Nachteil der Proteinbestimmung nach Bradford ist, dass die Stabilisierung der Sulfonatform von Coomassie Brillantblue G-250 vor allem durch oben erwähnte Aminosäuren bewirkt wird und die Empfindlichkeit sich somit von Protein zu Protein stark ändern kann (abhängig von der Primärstruktur des Proteins).
Bei einer großen Zahl organischer Verbindungen kann man nach Einstrahlung in deren Absorptionsbande spontane Emission von Licht beobachten.
Fluoreszenz: (stark vereinfachte Darstellung, ausführlicher siehe Analytische Chemie III)
Absorption von Energie ist für die Anregung von Elektronen aus deren Grundzustand (S0) in einen angeregten Zustand (>S0) verantwortlich. Nach erfolgter Anregung fallen die Elektronen umgehend (innerhalb von Picosekunden) in den für sie energetisch günstigeren Anregungszustand (S1), aus dem sie anschliessend über unterschiedliche Mechanismen weiter Energie abgeben, um wieder den Grundzustand zu erreichen (Jablonski Diagramm):
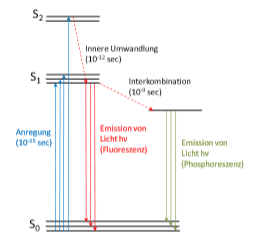
Mögliche Mechanismen der Energiekonversion:
Absorptionsspektren und Fuoreszenzspektren sind in der Regel verschoben, da der Energieverlust nach dem Anregungsprozeß zu einer Verschiebung der Emissionsstrahlung zu größeren Wellenlängen führt.
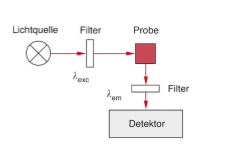
Im einfachsten Fall wird die Probe bei einer festen Wellenlänge (λexc) angeregt und die Intensität der Fluoreszenz bei fester Wellenlänge (λem) gemessen. Die beiden Wellenlängen werden durch Filter gewählt, die komplementär sein müssen. Werden sie durch durchstimmbare Filter (Monochromatoren) ersetzt, so können Emissionsspektren (λexc fest, Aufzeichnen der Fluoreszenzintensität als Funktion der Emissionswellenlänge λem) oder Aktionsspektren (λem fest, Aufzeichnen der Fluoreszenzintensität als Funktion der Anregungswellenlänge λexc) aufgenommen werden.
Fluoreszenzspektroskopie ist eine der weitverbreitesten spektroskopischen Methoden im bioanalytischen Bereich. Ohne Farbstoffzugabe kann nur die Fluoreszenz aromatischer Aminosäuren bei etwa λexc 280 nm angeregt und die Emission bei etwa λem 320-350 nm, je nach Protein, gemessen werden. Tryptophan (λexc 285 nm, λem 360 nm) Tyrosin (λexc 275 nm, λem 310 nm) Phenylalanin (λexc 260 nm, λem 283 nm)
Man spricht in diesem Fall von intrinsischer Fluoreszenz. Diese Methode ist für Proteinkonzentrationen von 5-50 µgmL-1 einsetzbar, aber in einem kleineren Konzentrationsbereich linear und stark abhängig von der Polarität des Lösungsmittels bzw. dessen pH Wert.
Der Beitrag von Phenylalanin zur Fluoreszenz ist nicht merklich, die Fluoreszenz von Tyrosin ist leicht auszulöschen (quenching), d.h. der Hauptbeitrag für diese Art der Fluoreszenzmessung erfolgt durch Tryptophan. Da Tryptophan in polarer Umgebung (Lösungsmittel) rotverschobene Fluoreszenz aufweist, kann über die Messung des Emissionsspektrums auf die molekulare Umgebung geschlossen werden.
Beispiel: Absorptionsspektren und Emissionsspektren von Lichtsammelpigmenten des photosynthetischen Bakteriums Rhodobacter capsulatus.
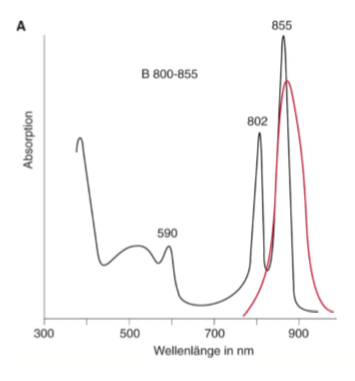
Absorptionsspektrum (schwarze Linie) des peripheren Bakteriochlorophyll-Protein-Komplexes mit zwei unterschiedlichen Pigmentpopulationen, die bei ca. 800 nm und bei ca. 850 nm absorbieren (B 800-850).
Wird dieser Komplex angeregt, so erfolgt ein schneller Energietransfer innerhalb der einzelnen Pigmente und zwischen B 800 und B 850, sodass die Fluoreszenz (rote Linie) nur vom niedrigstliegenden Übergang von B 850 beobachtet wird. Das Maximum der Fluoreszenz liegt bei ca. 865 nm.
Durch das Einbringen einer Fluoreszenzsonde durch chemische Reaktion oder Anlagerung kann extrinsische Fluoreszenz erzeugt werden.
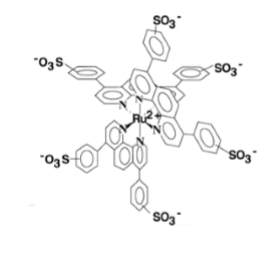
Ruthenium II-Bathophenanthrolin Disulfonat Chelat:
Reaktion mit Fluoraldehyd (OPA):
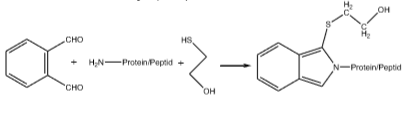
In der Gegenwart von Mercaptoethanol reagiert o-Phthalaldehyd (OPA) mit einem primären Amin (am Peptid/Protein) zu einem fluoreszenzgelabelten Analyten.
Reaktion mit Fluorescamin:
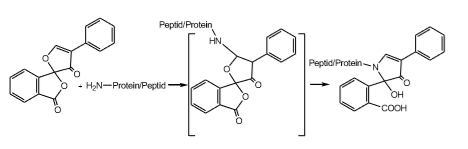
Fluorescamin reagiert mit primären Aminen und der Lysinseitenkette nahezu sofort bei Raumtemperatur und ermöglicht den Nachweis von Proteinkonzentrationen im Picomolbereich.
Reaktion mit NHS (N-Hydroxysuccinimid) – Ester Fluoreszierender Komponenten (Cy3, Cy5, o.ä.):
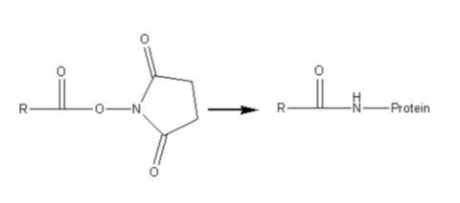
R = Rest, der „designed“ werden kann: Modulation der Anregungs- und Emissionswellenlängen je nach Anwendungsbereich (z.B. Laserinduzierte Fluoreszenz - LIF) durch den Einsatz unterschiedlich anregbarer Reste
Ein Analysenergebnis ist immer die Funktion eines Messwertes
x´ = f (y´)
und basiert auf der Anwendung der in einem Kalibrierexperiment gewonnen Kalibrierfunktion:
Messwert = Funktion des Substanzgehaltes y = f (x)
Löst man diese Gleichung nach x auf, wird die Kalibrierfunktion zur Erstellung der Analysenfunktion genutzt, die nach dem Einsetzen des Messwertes der untersuchten Probe das Analysenergebnis liefert.
Wahl des Arbeitsbereiches einer Kalibrierfunktion:
Die Ermittlung des Kalibrierfunktionstyps erfolgt im einfachsten Fall durch graphische Darstellung der Kalibrierdaten indem das Messsignal gegen die Analytkonzentration aufgetragen wird.
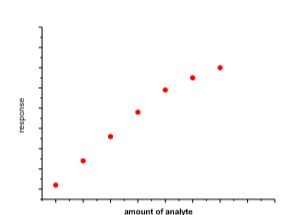
Hier liegt der Fall einer linearen Regression vor: y = a + bx; R2
Response = Anstieg ´ Konzentration + Achsenabschnitt
aber: letzten 2 Messsignale liegen ausserhalb des linearen Bereichs
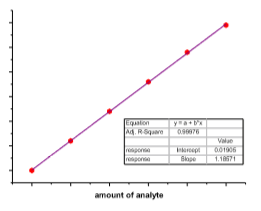
Die Linearität (y = a + bx) ist rechnerisch zu überprüfen und spiegelt sich in R2 (Korrelation) wieder.
Die Nachweisgrenze ist eine Entscheidungsgrenze für den qualitativen Nachweis des Analyten. Sie stellt den kleinsten Messwert dar, der mit einer vorgegebenen Sicherheit vom Blindwert zu unterscheiden ist. Zur praktischen Ermittlung wird meist die Standardabweichung des Blindwertes (mindestens 6 Blindwerte) herangezogen:
xN = Blindwert + Prognoseintervall für zukünftige Blindwerte
Die Erfassungsgrenze gibt den Mindestgehalt einer entsprechenden Probe an, der mit hinreichender Sicherheit nachgewiesen werden kann. Die Erfassungsgrenze entspricht dem 2fachen der Nachweisgrenze: xE = 2 * xN
Die Bestimmungsgrenze ist die kleinste Menge Gehalt einer Probe, die bei vorgegebener statistischer Sicherheit und maximal zugelassener relativer Abweichung (relativer Vertrauensbereich) quantitativ bestimmbar ist. Der Wert der Bestimmungsgrenze ist damit abhängig vom größten zufälligen Fehler.
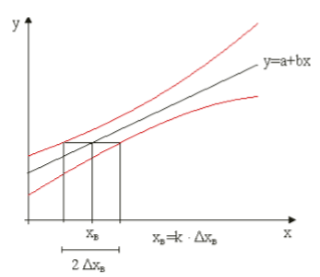
xB = 3 * xN (als Näherungswert)
t ... Student Faktor s0 ... Standardabweichung VB rel ... relative Vertrauensbereich
Die Angabe der xB muss immer im Zusammenhang mit dem VBrel Wert erfolgen !
Die Kalibrationsempfindlichkeit drückt sich mathematisch in der Steigung der Kalibrierfunktion aus.
Die analytische Empfindlichkeit drückt sich mathematisch durch den Quotienten des Anstiegs der Kalibrierfunktion und der Standardabweichung des Messsignals aus.

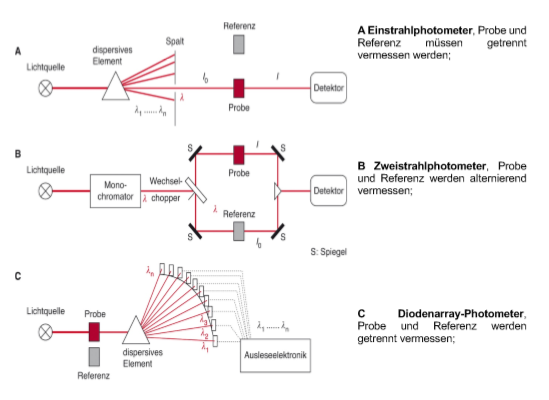
Methode A: Aufnahme eines Absorptionsspektrums

Methode B: Messen der Absorption einer Probe

Ausstattung – 3 LEDs: Das Fluoreszenzphotometer ist mit 3 LEDs ausgestattet, die für unterschiedliche Fluoreszenzanwendungen geeignet sind. Für die Vermessung von Fluorescamin gelabelten Proteinen eignet sich das UV LED.
Lichtweg:
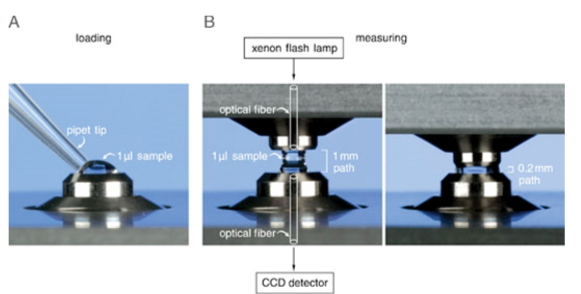
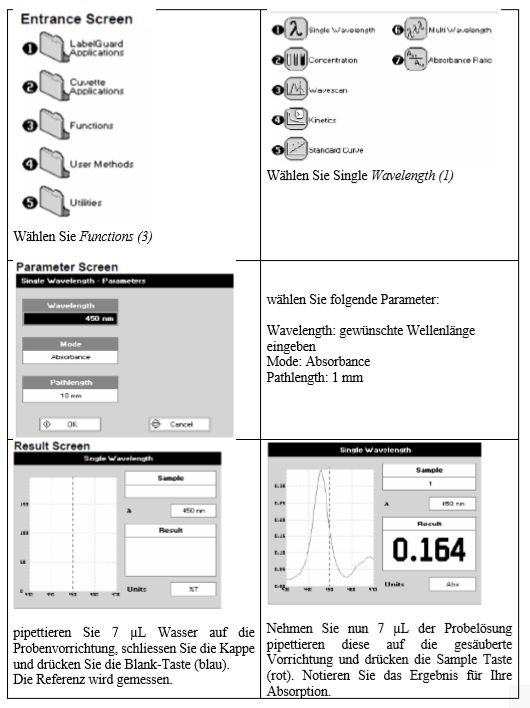
Nachdem man 2 µL Probe auf den unteren Probenteller pipettiert hat, wird der Deckel des Gerätes geschlossen und über die Software die Messung initiert.
Methode C: Aufnahme von Fluoreszenzemission (RFU = Relative Fluorescence Unit)
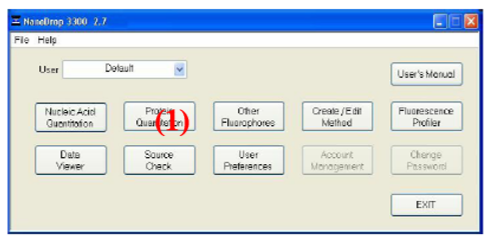
Auf der Bedienungssoftware Nanodrop 3300 wird der Button Protein Quantitation (1) äusgewählt und Fluorescamin als Methode gewählt.
Anschließend werden zur Initialisierung des Gerätes 2 µL Wasser auf den Probenteller pipettiert und der Arm geschlossen.
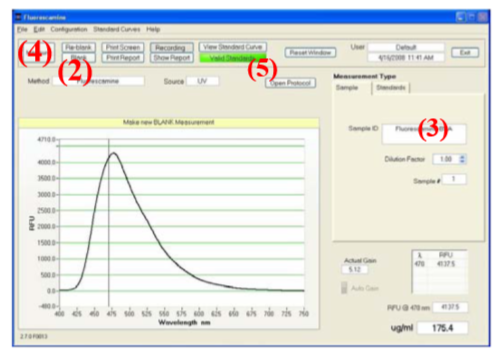
Die Initialisierung erfolgt durch drücken des Button Blank (2). Nach der Initialisierung wird der Wassertropfen vom Probenteller mit dem dort vorhandenen Kleenex-tuch abgewischt.
Nachdem man 2 µL Probe auf den sauberen Proben-teller pipettiert hat schreibt man die Probenbezeichnung in das Feld Sample ID (3) und drückt Measure (4).
Danach wischt man die Probe ab, pipettiert die nächste Probe, gibt die Probenbeziechnung an und misst. Jede einzelne Probe wird 3x gemessen.
Sobald alle Standards und die Proben gemessen wurden, wählt man Show Report (5) und „Save Report as ...“ aus dem Dropdown Menü:
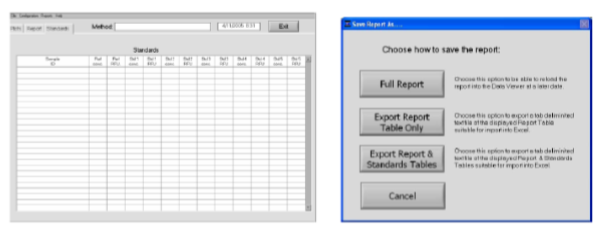
„Export Report Table Only“ ermöglicht es, die erhaltene Messwerttabelle als *.txt abzuspeichern, anschliessend zB in Excel zu importieren und dort eine Kalibrierfunktion zu erstellen.
ACHTUNG: Alle Glasgefäße vorher gut reinigen, da Sie nicht wissen wie gut Ihr Vorgänger diese gereinigt hat (Proteinspuren).
Für die Probenvorbereitung und die Verdünnungsreihe wird PBS-Puffer (Phosphate Buffered Saline pH 7,4) verwendet (bereitgestellt)
PBS-Puffer: (Laboranten/Tutoren geben Lösung aus – 20mL je Gruppe) 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4 und 0.27 g KH2PO4 werden in 800 mL dest. Wasser gelöst, mit NaOH oder HCl der pH auf 7.4 gebracht und anschliessend wird auf 1 L aufgefüllt.
Die ausgegebene Probe unbekannter Menge wird mit PBS auf 1 mL aufgefüllt und der ausgegebene Standard, gamma Globulin, wird in 1.5 mL PBS gelöst. Damit haben Sie eine 5 mg/mL gG-Stammlösung hergestellt (vorsichtig - Probe schäumt; nicht korrekt aufgefüllte Proben (verspritzen) werden NICHT erneut ausgegeben) Nach dem Zupipettieren des PBS Puffers das Reaktionsgefäß gut über Kopf schütteln, um sicherzustellen, dass der gesamte Feststoffanteil (auch wenn dieser nicht klar sichtbar ist) im Puffer gelöst ist.
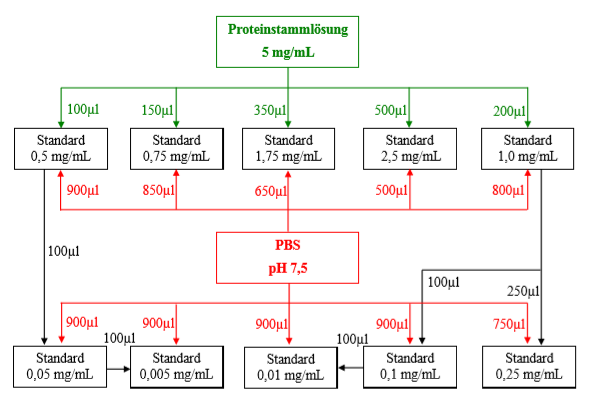
Durch Verdünnen mit PBS werden folgende Proteinkonzentrationen hergestellt:
Alle Standards können in entsprechenden Reaktionsgefäßen mit den Volumen 0,5 bzw 1,0 mL hergestellt werden. ACHTUNG: Standard 1,0 mg/mL muss entsprechend portiniert werden.
Pipertierschema:
Jede der Proteinlösungen (insgesamt 13: Proteinstammlösung, 10 Standards, Blindwert, unbekannte Probe) wird nach jeder der genannten photometrischen Methoden bestimmt. Die daraus resultierenden Kalibrierfunktionen werden gezeichnet. Geben Sie bei den Grafiken immer alle Messwerte (entweder einzeln oder als Mittelwert + Standardabweichung) an, auch wenn diese nicht für die Kalibrierfunktion relevant sind. Markieren Sie Werte, die Sie nicht zur Auswertung heranziehen farblich. Fügen Sie die Kalibrierfunktion bei den Werten ein, die Sie zur Erstellung der Geraden herangezogen haben.
Die Konzentration der Probe wird an Hand der am besten geeignet erscheinenden Methode bestimmt.
Ebenso soll der molare Extinktionskoeffizient jeder Bestimmungsmethode bei einer Konzentration von 1 und 0.1 mg mL-1 berechnet werden und die Kenngrößen der Kalibrierfunktion ermittelt werden (xN, xE, xB, (linearer) dynamischer Bereich). Standard
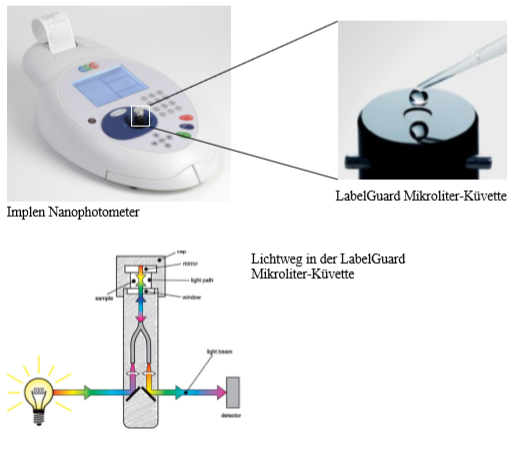
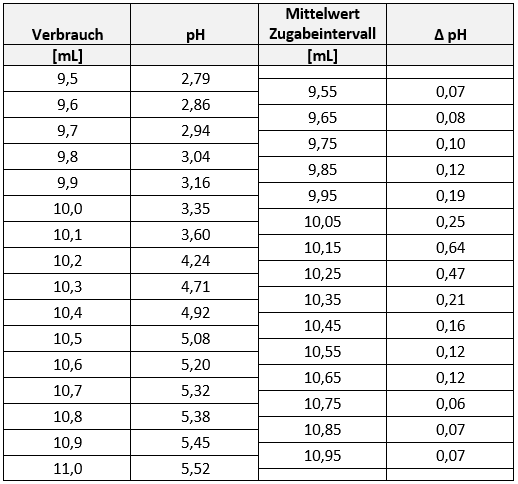
Um eine ausreichend gute Statistik für xN zu bekommen, messen Sie bitte 6 Blindwerte je photometrischem Experiment. Daraus können Sie den Vertrauenbereich errechnen. Der Blindwert einer Messung ergibt sich aus der PBS-Lösung und den entsprechenden Reagentien. Das UV/Vis Photometer verlangt nach einer Referenzmessung bevor die Absorption der einzelnen Lösungen gemessen werden können. Diese Referenz ist in allen Fällen destilliertes Wasser. (Gerätedetails und Messdetails siehe Geräte/Implen Nanophotometer).
Mit Methode A wird das Absorptionsspektrum der Standardlösung (1 mg/mL) aufgenommen und jenes Absorptionsmaximum bestimmt, welches für die Proteinquantifizierung am geeignetsten erscheint
Die Absorbtion der hergestellten Proteinlösungen werden bei der zuvor bestimmten Wellenlänge am Nanophotometer mit Methode B gemessen. Dazu werden jeweils 7 µL der Blindwerte, der Standardlösungen und der Probelösung auf die LabelGurad Küvette pipettiert.
| Lösung A: | 0,1M Natronlauge (NaOH) (bei Laboranten/Tutoren) |
| (ca. 3-5 mL in einem 5 mL Becherglas) | |
| Lösung B: | 4 % Natriumcarbonat (Na2CO3 ´ 10H2O) in Wasser (bei Laboranten/Tutoren) |
| (ca. 3-5 mL in einem 5 mL Becherglas) | |
| Lösung C: | 1 % Kupfersulfat (CuSO4*5H2O) in Wasser (bei Laboranten/Tutoren) |
| (ca. 1 mL, in einem 5 mL Becherglas) | |
| Lösung D: | 2 % Na-K-Tartrat in Wasser (bei Laboranten/Tutoren) |
| (ca. 1 mL in einem 5 mL Becherglas) |
| Lowry Reagens A (frisch zubereiten !!!) | In einem 5 mL Becherglas werden 2.45 mL Lösung A + 2.45 mL Lösung B gemischt, danach 50 µL Lösung C zugeben, danach 100 µL Lösung D zugeben. |
| Lowry Reagens B Folin Ciocalteu abholen bei Laboranten/Tutoren | In einem weiteren 5 mL Becherglas werden 1 mL Folin-Ciocalteu Phenol Reagenz mit 1 mL dest. Wasser gemischt |
50 µL Probe (Protein unbekannter Konzentration, Proteinstandardreihe und auch PBS) werden mit 250 µL Lowry Reagenz A versetzt, gut geschüttelt und 10 min bei Raumtemperatur stehengelassen. Danach gibt man rasch 25 µL Lowry Reagenz B zu, schüttelt gut und lässt diese Mischung 30 min bei Raumtemperatur inkubieren. Die Vermessung erfolgt bei 750 nm mit Methode B.
Bei allen Messungen ist darauf zu achten, dass der Zeitraum zwischen dem Mischen der Proteinlösung und der Vermessung in etwa gleich groß ist.
| 3 mg/mL Fluorescamine Ausgabe | geben Sie 1 mL Aceton (beim Laboranten/Tutoren) in das Eppendorfgefäß mit dem Feststoff |
150 µL Probelösung (Protein unbekannter Konzentration, Proteinstandardreihe und auch PBS) werden mit 50 µL Fluorescamine Lösung gut vermischt und nach 5 min die Emission bei 470 nm mit Methode C vermessen. (Aceton ist leicht flüchtig – halten Sie das Gefäß mit dem Derivatisierungsreagens nicht in der warmen Hand, da sich die Konzentration durch die Verflüchtigung des Lösungsmittels stark ändert)
Tragen Sie für jede photometrische Bestimmung die gemessenen Absorptions- bzw. Emissionswerte gegen die hergestellten Proteinkonzentrationen auf. Beurteilen Sie visuell, welcher Bereich der Kalibrierfunktion linear ist. Berechnen Sie anschliessend die Kalibrierfunktion und dessen Korrelation für diesen Bereich.
Aus den gemessenen Blindwerten (6 unabhängige Messwerte) können Sie die Nachweisgrenzen, Bestimmungsgrenzen und Erfassungsgrenzen für jede Proteinbestimmungsmethode ermitteln.
Die molaren Extinktionskoeffizienten ermitteln Sie als Mittelwert aus 3 Einzelmessungen. Dazu ziehen Sie 3 unterschiedliche Konzentrationen aus der Mitte des linearen Kalibrationsbereichs jeder photometrischen Bestimmung heran.
Sie haben Ihre Probe mit allen vorgegebenen Proteinbestimmungsmethoden gemessen. Für die Auswertung (d.h. die Bestimmung des Proteingehalts Ihrer Probe) verwenden Sie nur solche Kalibrierfunktionen, die eine ausreichende Empfindlichkeit und Linearität für Ihre Probe haben. Vergleichen Sie dazu den Absorptions-/Emissionswert Ihrer Probe mit der Kalibrierfunktion – Wo liegt meine Probe auf der Geraden?
Für den Fall, dass mehr als eine Kalibrierfunktion für die Konzentrationsermittlung geeignet erscheint, vergleichen Sie die Ergebnisse (mg/mL) für Ihre Probe und geben bei guter Übereinstimmung einen Mittelwert (inkl. Vertrauensbereich) ab oder wählen Sie eine Methode, die Ihnen am sinnvollsten erscheint. In letzterem Fall ist unbedingt eine Begründung im Protokoll anzugeben!
Für die Berechnung des molaren Extinktionskoeffizienten müssen Sie die Proteinkonzentrationen in der Probelösung berücksichtigen (durch Zusatz der Reagenzien werden die Proteinlösungen teilweise weiter verdünnt und somit ändert sich die Konzentration).